
The diseases in this group either affect the retina in isolation or are associated with additional systemic manifestations. They are grouped because they are heritable and result in characteristic retinal findings. A good way to learn these conditions is to associate them with their retinal appearance.
Retinitis Pigmentosa (RP)
RP is the commonest inherited retinal disorder. It is characterised by generalised photoreceptor dysfunction (rods first, then cones) and progressive atrophy of the retina.
Genetics
- Inheritance is most commonly AD (mildest) but can be AR or XL (worst).
- Commonest genes for AD → RHO, RP1, PRPF31
- Commonest gene for AR → USH2A
- Commonest gene for XL → RPGR
RP is most commonly caused by a rhodopsin gene mutation on chromosome 3
Diagnostics
Presentation
- Nyctalopia + tunnel vision + decreased visual acuity
- Associated with posterior subcapsular cataract, CMO, open-angle glaucoma and keratoconus
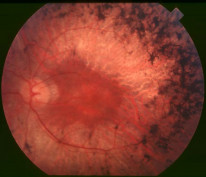
A fundus photograph of a patient with retinitis pigmentosa. By Christian Hamel, CC BY 2.0 , via Wikimedia Commons.
Investigations
- A triad of fundoscopic findings: waxy disc + bony spicules + arteriolar attenuation
- Optic disc drusen may be seen
- ERG is electronegative - this is a helpful diagnostic finding and can also be used to monitor disease progression.
- EOG is also abnormal because of global photoreceptor RPE dysfunction
Initially, scotopic ERG (ERG in the dark) is worse than photopic (in the light) because rods are affected first.
Management
- No specific treatment is available as of yet but novel genetic therapies have shown promise in early clinical trials
RP Associated Conditions
RP associated conditions are a specific group of disorders which are all characterised by photoreceptor dysfunction, nyctalopia and tunnel vision. They can be thought of as variants of RP.
Diseases |
Key characteristics |
|---|---|
Usher syndrome |
Commonest inherited disorder of combined sensorineural deafness and blindness |
Refsum syndrome |
Accumulation of phytanic acid leading to: Anosmia + peripheral neuropathy + ichthyosis |
Bardet-Biedl syndrome |
Learning disability + polydactyly + obesity |
Bassen Kornzweig syndrome |
Abnormal absorption of fat-soluble vitamins leading to spinocerebellar ataxia and acanthocytosis |
The RP associated conditions are inherited in an autosomal recessive fashion.
Leber Congenital Amaurosis
Leber congenital amaurosis is a genetic cause of visual impairment in children. It is characterised by generalised dysfunction of photoreceptors.
Pathology
- AR severe rod-cone dystrophy.
- Associated with multiple systemic features such as learning difficulty and epilepsy
Diagnostics
Presentation
- Blindness at birth + nystagmus + absent pupil reflexes
- Early disease → normal fundus
- Late disease → Salt and pepper retinopathy + bulls-eye maculopathy
Investigations
- ERG is non-recordable because the photoreceptors are not working
Best Disease
Best vitelliform macular dystrophy is the second commonest inherited macular dystrophy. It is characterised by a classic 'egg yolk' appearance of the macula.
Pathology
- AD mutation of the bestrophin gene on Chr11q13 → channelopathy of the RPE → abnormal lipofuscin accumulation and photoreceptor atrophy
Diagnostics
Presentations
- Bilateral egg yolk macula (yellow circular elevated fundus lesion)
Investigations
- Characterised by an egg yolk macula on fundoscopy
- ERG → Normal
- EOG → abnormal with reduced Arden ratio
This is the classic cause of normal ERG with abnormal EOG.
Stargardt Disease
Stargardt's disease is the commonest inherited macular dystrophy and typically presents in childhood.
Pathology
- AR mutation of ABCA4 on Chr1 → diffuse accumulation of lipofuscin in the RPE
Diagnostics
Presentation
- Presentation: reading difficulties in teenagers
Investigations
- The fundus can appear normal in the early stage.
- As the disease progresses, fundoscopy will show a beaten bronze macula with diffuse yellow specks
- FFA shows a classically dark choroid due to blockage of fluorescence by lipofuscin
Albinism
Albinism is the deficiency of melanin pigment. It can affect the eye in isolation (ocular albinism) or it can be systemic (oculocutaneous albinism).
Pathology
- Oculocutaneous albinism (AR) is more common than ocular albinism (XL)
- Decreased visual acuity is typically due to foveal hypoplasia
Pigmentation is useful in ocular structures because it allows the careful filtering of light. In ocular albinism, the iris and RPE are hypopigmented. This can lead to photophobia because too much light is bouncing around inside the eye.
Diagnostics
Presentation
- Presentation: dVA (foveal hypoplasia) + nystagmus + strabismus + iris hypopigmentation

(A) is a fundus in a patient with albinism. (B) is a normal fundus. By Karen Grønskov CC BY 2.
Interestingly, patients with albinism have been found to have an increased decussation of temporal retinal fibres at the optic chiasm, on visual evoked potentials
Management
- From an ocular perspective, the main priority is to treat the ametropia.